Back in 2015, the discovery of genomic mutations led to the suspension of the first ever human trial using Pluripotent Stem Cells (PSCs) at the RIKEN Institute in Japan (Garber, 2015). These genetic alterations had not been detected in the original patient’s fibroblast, but might have developed in culture.
Fortunately, they were spotted before injection to the patient, but a lesson was learned. CNVs (Copy Number Variations) accounted for half of the detected genomic defects identified in the hPSC-derived products used in this clinical trial. The impact they had was a clear signal that CNVs needed to be closely monitored using appropriate technological detection methods. Let’s take a closer look at them, explain why they matter to you as a research scientist working on PSCs, and explore solutions to help you detect them in time.
What are CNVs?
Copy Number Variations are deletions or duplications of chromosomal segments that can be found in 12% of the human genome (Redon et al., 2006). They are therefore widespread in the human population, and fortunately most of them are benign variants that will not cause diseases. Small differences in phenotypes are regarded as normal variations, especially when common in a population with small changes in physiology (benign traits or common diseases and intolerances). They reflect human evolution. Larger deviations however have been found to be an underlying factor in many diseases. Recent reviews have found that 17 conditions of the nervous system alone including Parkinson’s and Alzheimer’s diseases can result from CNVs (Source: www.gene-quantification.de).
The most frequent type of abnormalities detected in pluripotent stem cells are CNVs!
Back in 2020, Dr. Said Assou, Professor John de Vos and their team undertook the mammoth task of exhaustively cataloguing genetic structural variants previously detected in hPSCs. They collected data on 942 cell samples from 107 different studies published between 2004 and 2016.
They found that the most recurrent abnormalities were CNVs. They then plotted the genomic coordinates of these 738 identified recurrent abnormalities including the known hotspots of chromosomes 1, 12, 17q, 20q11.21 or X (Assou et al., 2020). They subsequently designed 24 probes that covered 90% of the 738 recurrent abnormalities detected. These form the foundations of our company, Stem Genomics.
But should CNVs really be a cause for concern among scientists working on PSCs?
Beyond their potential impact on human health in general, CNVs have been known to alter the differentiation propensity of PSCs in culture (Keller & Spits, 2021). Keller et al. observed the impact of gains of 12p and 20q, two of the most recurrent CNVs in hPSCs, on differentiation. These mutations led to a general reduction in differentiation capacity in cells carrying a 12p13.31 gain (Keller & Spits, 2021). Markouli et al. also demonstrated that gains of 20q11.21 caused a near complete loss of neurectoderm differentiation capacity as a result of altered TGF-beta and SMAD signaling (Markouli et al., 2019).
So what causes CNVs in culture?
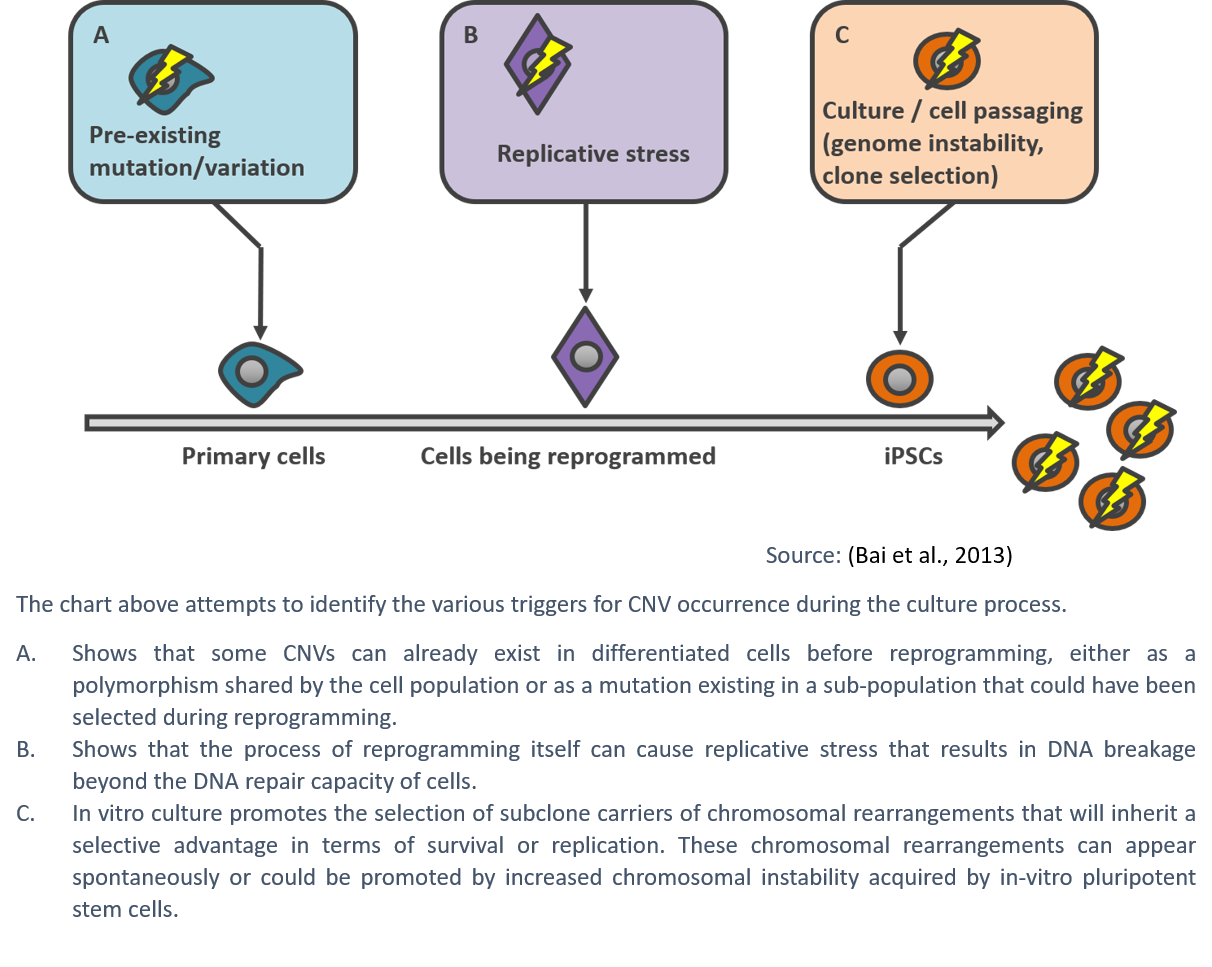
The mechanisms driving the high prevalence of chromosomal abnormalities in hPSCs are still poorly understood, but are generally thought to be the result of suboptimal culture techniques and a high level of damage compared to somatic cells. As human pluripotent stem cells proliferate rapidly, they are susceptible to replication stress, which can lead to DNA damage and instability resulting in karyotypic abnormalities (Keller & Spits, 2021).
Although improvements in culture methods, such as the use of low oxygen conditions or the addition of nucleosides in the culture media, can help alleviate the issue (Halliwell et al., 2020; Thompson et al., 2020), the safe approach remains regular testing.
In practice: how can you detect CNVs?
CNVs can be detected using various technologies, namely G-Banding, FISH, qPCR, dPCR or NGS. While they all present advantages and disadvantages, Dr. Said Assou and his team selected digital PCR for the high precision detection this technology provides at a reasonable cost (Assou et al., 2018). Digital PCR is a targeted method that relies on the knowledge of specific positions – in this case recurrent mutations – for primer and probe design. Therefore, the team undertook further research to maximize the detection of recurrent abnormalities using a limited number of probe assays.
In their paper published in Nature, Hastings et al. highlighted the fact that CNVs form at a higher rate than any other types of mutations (Hastings et al., 2009).
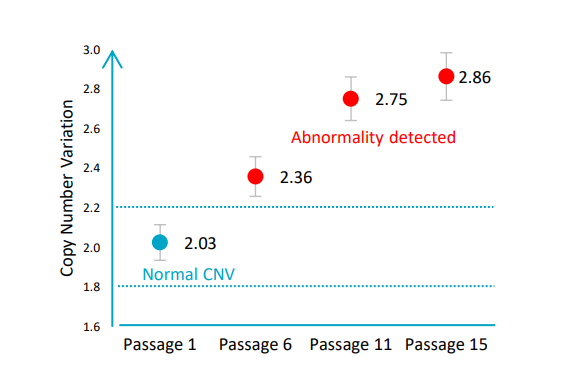
This was also described by Lauer et al. when they observed that copy number variants arise early and repeatedly, that they are diverse in size and copy number, and that they are generated at a high rate (Lauer et al., 2018). In the specific case of hPSCs, Dr. Assou and his team found that a genetically normal hiPSC line can acquire harmful CNVs in five passages (Assou et al., 2020). This was also previously observed by other research scientists looking at PSCs (McIntire et al., 2020; Pamies et al., 2017).
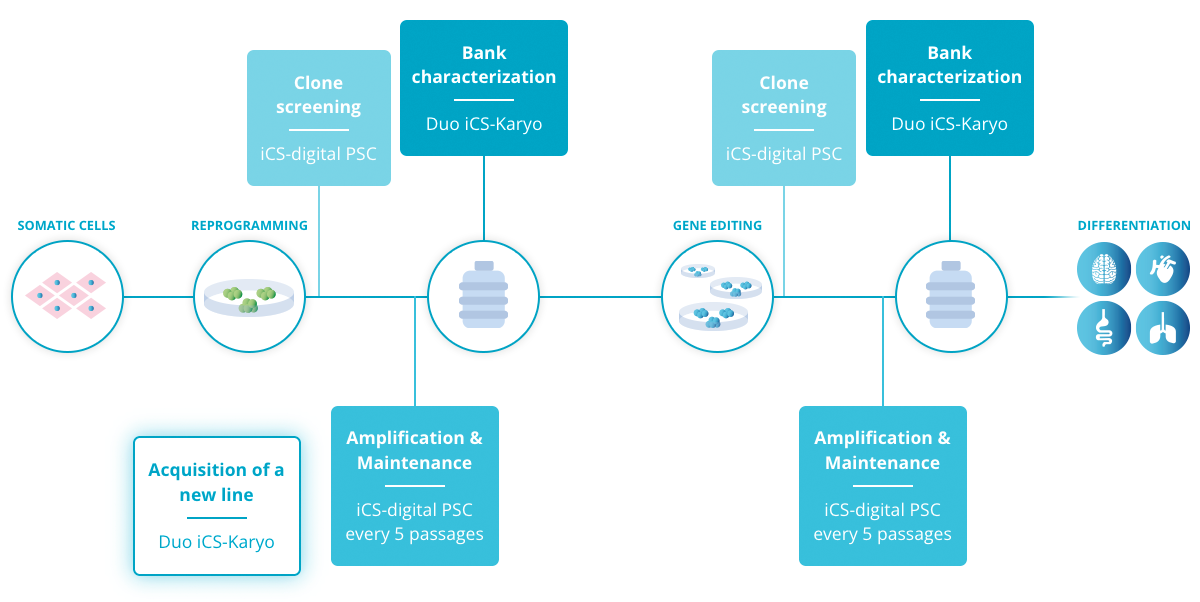
All things considered, these results support the use of regular in-process tests to detect abnormalities as early as possible and limit the impact on the cell culture. As a good practice, we recommend testing during the following stages: during cell amplification and maintenance, at clone screening and at pre-banking characterization (Read more).
Consequently, Stem Genomics launched the iCS-digital™ PSC as a service and later on, as a kit, to enable PSC researchers to accurately and rapidly detect the most recurrent CNVs in hPSCs in a cost-effective way.